Case Report - (2015) Volume 4, Issue 4
Multiple sclerosis (MS) is very similar to the Gougerot Sjögren Syndrome (GSS) by its clinical presentations. Features found in common between the two disorders include symptoms of lesions of the brain, spinal cord and optic tract, the detection of autoantibodies, such as antinuclear, anti-Ro, anti-La and the prevalence of MRI abnormalities such as periventricular and subcortical lesions. Almost all symptoms found in patients with the central nervous system (CNS) -GSS involvement described above could be attributed to the concomitant presence of MS. Therefore, the differential diagnosis of these diseases is difficult. The authors present the case of a 26 years old woman who presented a GSS with neurological manifestations mimicking MS.
Keywords: Gougerot sjögren syndrome; Neurological manifestations; Multiple sclerosis; Diagnosis; Treatment
Neurologic involvement occurs in approximately 20-25% of patients with GSS [1,2]. Manifestations are diverse and can affect the entire neuroaxis. CNS dysfunction involves the brain as well as the spinal cord and may recur over time. Due to a variety of presentations, GSS with neurologic involvement may be difficult to diagnose and may be particularly challenging to distinguish from MS, especially during initial presentation.
The relationship between MS and GSS is ambiguous; it was suggested that some patients diagnosed with MS may instead have GSS.
A 26-year-old woman without personal history of sicca syndrome (xerophthalmia and/or xerostomia) was presented in August 2009 with vertical diplopia and a drop of the left eyelid regressing spontaneously after 2 months. She developed 3 months later after the diplopia a heavy right hemibody with incomplete improvement. She consulted in March 2012 for aggravated heaviness of right hemibody. Neurological examination had objectivated quadripyramidal syndrome, static and kinetic cerebellar syndrome and later cordonal syndrome. Spinal cerebral MRI had objectivated lesions in hyperintense T2 and FLAIR in the callosum corpus with radial ridges in hull (Figure 1) and in periventricular white matter, in the semi oval center and juxtacortical lesions (Figure 2).
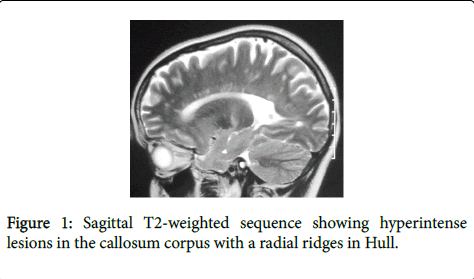
Figure 1: Sagittal T2-weighted sequence showing hyperintense lesions in the callosum corpus with a radial ridges in Hull.
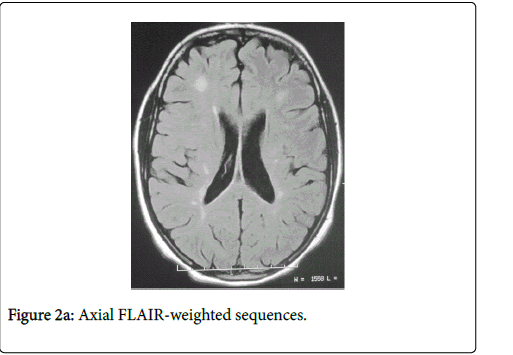
Figure 2a: Axial FLAIR-weighted sequences.
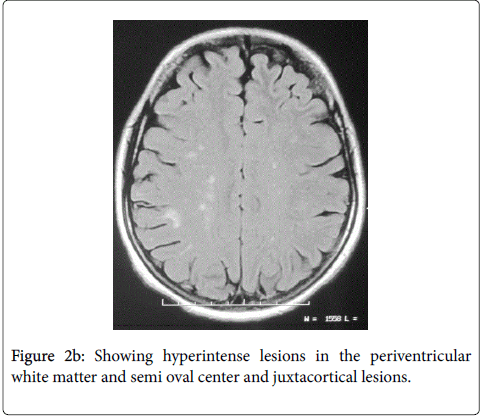
Figure 2b: Showing hyperintense lesions in the periventricular white matter and semi oval center and juxtacortical lesions.

Figure 3: A lesion of the frontal white matter is taking active gadolinium.
A lesion of the frontal white matter took gadolinium (Figure 3). Spinal MRI was normal. Visual evoked potentials (VEP) were in favor of bilateral and asymmetric demyelinating optic neuropathy predominant on the left side. The study of cerebrospinal fluid (CSF) was in favor of intrathecal synthesis of IgG. The electromyogram was normal. Blood count showed lymphopenia at 1200 elmts / mm3.
The erythrocyte sedimentation rate was increased to 117 mm the first hour. A series of further investigations was undertaken. These diagnostic tests were directed by the principal diagnoses initially mentioned: MS, sarcoidosis, vasculitis, systemic lupus erythematosus and GSS. Renal, hepatic, calciun and phosphate balances as well as the rate of angiotensin converting enzyme were normal. Chest radiography and chest CT scan showed no abnormalities. Immunological balance showed positive antinuclear antibody at 1/200 with anti-SSA antibodies and rheumatoid factor positive. The "Break up time" (BUT) was altered to 3'' in favor of xerophthalmia. Biopsy of salivary glands was in favor of a Chisholm stage IV sialadenitis. After this diagnostic approach, we attached this neurological manifestation to GSS. Nevertheless, MS is not excluded. The patient had a steroid bolus of 1 g for three days and then was put on orally corticosteroids at a dose of 1 mg/kg/day with a favorable outcome.
MS is the most common demyelinating disease caused by an autoimmune inflammatory process in the CNS and is associated with aberrant immune response to myelin selfantigens. Coexistence of MS with other autoimmune disorders, including GSS, systemic lupus erythematosus, rheumatoid arthritis and scleroderma has been reported previously.
GSS is a chronic autoimmune inflammatory disease of unknown etiology. The CNS symptoms are present in 20-25% of the patients with GSS [1,2]. Some patients have a relapsing-remitting course mimicking MS. However, all symptoms found in patients with the CNS-GSS involvement could be attributed to the concomitant presence of MS. Therefore, the differential diagnosis of these diseases is difficult.
Our patient presented a GSS with neurological manifestations. Two hypotheses have been suggested: the first is the explanation of the whole table by GSS mimicking MS and the second is that of a coexistence of GSS and MS.
Indeed, the GSS shares with MS several neurological disorders, confirming the first hypothesis. However, the second hypothesis is also possible to change by relapses and remissions and paraclinical explorations (MRI, EPI and CSF oligoclonal profile) in favor of MS. This last hypothesis was supported by some studies [3-5]. The prevalence of GSS in MS patients varies in the published literature (0-16.6%) [3].
In the study of Miró [4] sixty-four patients with clinically or laboratory-supported definite MS were evaluated for evidence of GSS, 2 patients (3.1%) were found with clinical evidence of GSS. Sandberg-Wollheim [5] concludes that MS and GSS may coexist in the same individual, but GSS is not more common among MS patients than expected in the general population.
Neurological manifestations can reveal the GSS. Thus, in the series of Alexander et al. on patients with central neurological manifestations, neurological manifestations preceded sicca syndrome in 12% of cases [6]. This makes the diagnosis of these neurological manifestations often difficult with a long delay in diagnosis.
In our case, the neurological manifestation was revealing with clinical outcome and paraclinical explorations (MRI, VEP and CSF oligoclonal profile) that can mimic MS relapsing.
Our patient was treated as GSS with neurological manifestations. However, MS is the main differential diagnosis, what motivates a regularly clinical and paraclinical (MRI) monitoring.
Indeed, in the study of Morgen et al. assessing the radiological criteria for MS in patients with GSS, Barkhof criteria were present in 39% of patients but with less involvement of the callosum corpus [7]. Lesions of the gray matter are also described, involving the basal ganglia and the cerebral cortex. The lesions are sometimes numerous and their size can exceed 10 mm. Contrast enhancement after gadolinium injection is rare [8].
CSF oligoclonal profile was found in 30% of patients with GSS with central nervous system impairment. This frequency is lower than in MS [9]. Cranial nerves may be affected in the GSS. The cranial nerve most frequently affected is the trigeminal (V) [10]. GSS can also be accompanied by a retrobulbar optic neuropathy (RBON) symptomatic or not. RBON frequency is variable from 12 to 61% of patients. The RBON can sometimes be bilateral [9,11]. In our case, the patient presented a bilateral RBON and an involvement of the common oculomotor nerve (III). Indeed, the oculomotor nerves can also be affected in the GSS with rarely impairment of the common oculomotor nerve [12]. Achieving of the common oculomotor nerve is also rare in MS, estimated at less than 1% [13].
Treatments of CNS -GSS involvement, usually comprise symptomatic approach in milder cases however; pulse cyclophosphamide and steroids or other immunosuppressants (chlorambucil or azathioprine) are required in cases with progressive symptoms leading to neurological impairment. Anti-TNF agents (infliximab and etanercept) and B cell targeted therapies (rituximab and epratuzumab) are used in primary GSS however their efficacy on CNS manifestation is still unclear [14]. In our case, oral corticosteroids at high doses, preceded by intravenous bolus was effective.
Neurological manifestations can reveal the GSS and can mimic MS but the coexistence is possible in the same individual. Whether this association is fortuitous or whether there is pathogenetic linkage between these two affections remains to be established.
