Journal of Multiple Sclerosis
ISSN - 2376-0389NLM - 101654564
Research Article - (2019) Volume 6, Issue 2
Multiple sclerosis (MS) is a progressive disease that is characterized by multifocal inflammation and demyelination in a central nervous system. Experimental autoimmune encephalomyelitis (EAE) is an animal model of MS that shows ascending flaccid paralysis with inflammation of spinal cord. We focus on the potential roles of inducible prostaglandin E2 (PGE2) and interleukin-1β (IL-1β) in EAE after myelin oligodendrocyte glycoprotein 35-55 peptide immunization in this review. PGE2 synthesized by cyclooxygenase (COX)-2 and microsomal prostaglandin E synthase-1 (mPGES-1) in vascular endothelial cells (VECs) or macrophages/microglia aggravates inflammation, demyelination and paralysis and facilitates the activation and differentiation of CD4-positive (CD4+) T cells into interleukin-17 (IL-17)-producing helper T cells to promote neuronal dysfunction and blood-spinal cord barrier disruption in the EAE model. PGE2 also causes vascularity and increases IL-1β production in VECs and CD4+ T cells, and IL-1β plays a crucial role in facilitating EAE progression and stimulates the synthesis of COX-2 and mPGES-1 to produce PGE2. Thus, the
local PGE2-IL-1ββ-PGE2 signalling pathway facilitates IL-17 production in inflammatory lesions in the spinal cord of EAE animals. This pathway represents a possible mechanism by which PGE2 participates in EAE pathology. Taken together, this evidence highlights the intercellular PGE2 signalling pathway in the spinal cord as a therapeutic target for ameliorating MS severity after disease onset.
Keywords: Prostaglandin E2; Interleukin-1β; Microsomal, prostaglandin synthetase-1; Multiple sclerosis; Experimental, autoimmune encephalomyelitis; CD4-positive (CD4+) T cells; Vascular, endothelial cells
MS: Multiple Sclerosis; PGE2: Prostaglandin E2; COX: Cyclooxygenase; mPGES-1: Microsomal Prostaglandin E Synthetase-1; mac/mic: Macrophages/microglia; VECs: Vascular Endothelial Cells; EAE: Experimental Allergic Encephalomyelitis; MOG35–55: Myelin Oligodendrocyte Glycoprotein35–55 Peptide; IL-1β: Interleukin-1β; IL-1RN: Interleukin-1 Receptor Antagonist; IL- 1R1: Interleukin-1 Receptor 1; mPGES-2: Microsomal Prostaglandin E Synthetase-2; cPGES: Cytosolic PGE2 Synthase; EP: E-prostanoid; mPGES-1-/-: Microsomal Prostaglandin Synthetase-1-deficient; Wt: Wild Type; IL-17: Interleukin-17; BBB: Blood-Brain Barrier; CD4+ T: Cell CD4-positive T cell; Th17: Interleukin-17-producing Helper T cell; Th1: Type 1 helper T cell; MMPs: Matrix metalloproteinases; VEGF: Vascular Endothelial Growth Factor; IL-17R: Receptor for IL-17; ASC: Apoptosis-associated Speck-like Protein Containing a Caspase Recruitment Domain.
Multiple sclerosis is a progressive disease showing multifocal inflammation and demyelination. Inducible prostaglandin E2 (PGE2) is an inflammatory mediator synthesized by cyclooxygenase (COX)-2 and microsomal prostaglandin E synthase-1 (mPGES-1, also known as PTGES). COX-2 is increased in macrophages/microglia (mac/mic) in the brains of MS patients and in vascular endothelial cells (VECs) in animals with experimental autoimmune encephalomyelitis (EAE), an animal model of MS [1,2]. In addition, mPGES-1 expression is induced in brain VECs in fever and neuronal injury after kainic acid injection [3,4]. Rodents with EAE induced by the myelin oligodendrocyte glycoprotein 35-55 peptide (MOG35–55) exhibit typical perivascular infiltration of mononuclear cells and inflammatory foci in the spinal cord and brain. MOG35–55-induced EAE also results in infiltration of the cerebral meninges at the third and lateral ventricles, as well as severe parenchymal infiltration in the spinal cord [5,6]. The symptomatic course of EAE involves progressive flaccid paralysis with inflammation which targets the spinal cord but accordingly, we mainly focus on spinal inflammation in EAE and its association with PGE2 in this review [7,8].
PGE2 is increased after EAE induction in spinal cord and treatment with selective COX-2 inhibitors prevent the development of EAE-associated paralysis. Similarly, the induction of mPGES-1 expression in infiltrating macrophages stimulates the clinical EAE progression in mice. Moreover, mPGES-1 expression is also induced in VECs located around inflammatory foci and accelerates inflammation, demyelination, and paralysis in EAE models. Thus, PGE2 produced by mPGES-1 promotes disease progression in the spinal cord of animals with EAE [9-15].
Interleukin-1β (IL-1β) is also an important inflammatory and pathological mediator of EAE mice. The administration of a recombinant interleukin-1 receptor antagonist (IL-1RN) delays disease onset and decreases EAE severity [16]. Additionally, the expression of a defective interleukin-1 receptor 1 (IL-1R1) gene in mice show complete resistance to EAE [17]. IL-1β is generally known to stimulate PGE2 production; whereas, evidence showing that PGE2 stimulates IL- 1β production is unavailable. However, we recently find that IL-1β is a mediator or component of the mechanism by which PGE2 promotes the progression of EAE [18].
In this review article, we summarize the roles of PGE2 and IL-1β in EAE and their relations with inflammatory molecules related to spinal cord inflammation. Furthermore, we discuss intercellular interactions among VECs, CD4+ T cells, and monocytes mediated by PGE2 and autocrine IL-1β signaling in EAE mice.
Patients with definite MS have higher baseline PGE expression in leukocyte cultures than healthy control subjects and PGE2 expression is similarly elevated in peripheral blood monocytes from chronic MS patients [19-20]. Based on these reports, peripheral PGE2 expression might be related to MS progression. PGE2 is synthesized from arachidonic acid by COX and PGES, and COX exists as constitutively active (COX-1) and inducible (COX-2) isoforms. MS patients show COX-2 induction in chronic, active lesions in the brain [1]. COX-2 expression also appears in mac/mic in the spinal cord 16 days after EAE induction [21]. In EAE models, COX-2 is localized to ECs in the spinal cord, particularly 14-25 days after immunization [2]. Thus, COX-2 in central nervous system (CNS) is a crucial mediator of MS and EAE pathology. PGE2 concentration is elevated in the spinal cord after EAE induction (Figure 1) [12].
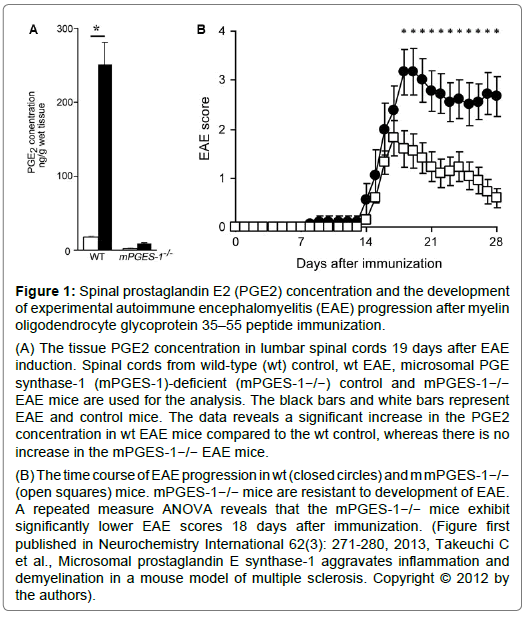
Figure 1: Spinal prostaglandin E2 (PGE2) concentration and the development of experimental autoimmune encephalomyelitis (EAE) progression after myelin oligodendrocyte glycoprotein 35–55 peptide immunization.
(A) The tissue PGE2 concentration in lumbar spinal cords 19 days after EAE induction. Spinal cords from wild-type (wt) control, wt EAE, microsomal PGE synthase-1 (mPGES-1)-deficient (mPGES-1−/−) control and mPGES-1−/− EAE mice are used for the analysis. The black bars and white bars represent EAE and control mice. The data reveals a significant increase in the PGE2 concentration in wt EAE mice compared to the wt control, whereas there is no increase in the mPGES-1−/− EAE mice.
(B) The time course of EAE progression in wt (closed circles) and m mPGES-1−/− (open squares) mice. mPGES-1−/− mice are resistant to development of EAE. A repeated measure ANOVA reveals that the mPGES-1−/− mice exhibit significantly lower EAE scores 18 days after immunization. (Figure first published in Neurochemistry International 62(3): 271-280, 2013, Takeuchi C et al., Microsomal prostaglandin E synthase-1 aggravates inflammation and demyelination in a mouse model of multiple sclerosis. Copyright © 2012 by the authors).
Three isoforms of PGES have been identified: mPGES-1, microsomal prostaglandin E synthase-2 (mPGES-2) and cytosolic PGE2 synthase (cPGES). Notably, mPGES-2 and cPGES are constitutively appeared, while mPGES-1 expression is induced in a manner functionally coupled to COX-2 in macrophages or osteoblasts following pro-inflammatory stimulation [22-25]. In the brain, mPGES-1 expression is induced by COX-2 in VECs during fever, kainic acid-induced neuronal injury, and cerebral ischemia [3,4,26]. In EAE models, mPGES-1 is expressed in ECs macrophages and mac/mic (Figure 2) but not in CD4+ T cells in the spinal cord [11,12,18]. COX-2 expression is also induced in ECs, but conflicting information about the induction of COX-2 expression in mac/mic has been reported [2,21]. Therefore, VECs in CNS are undoubtedly important sources of PGE2 synthesized by COX-2 and mPGES-1 in response to pro-inflammatory changes in the spinal cord of EAE animals.
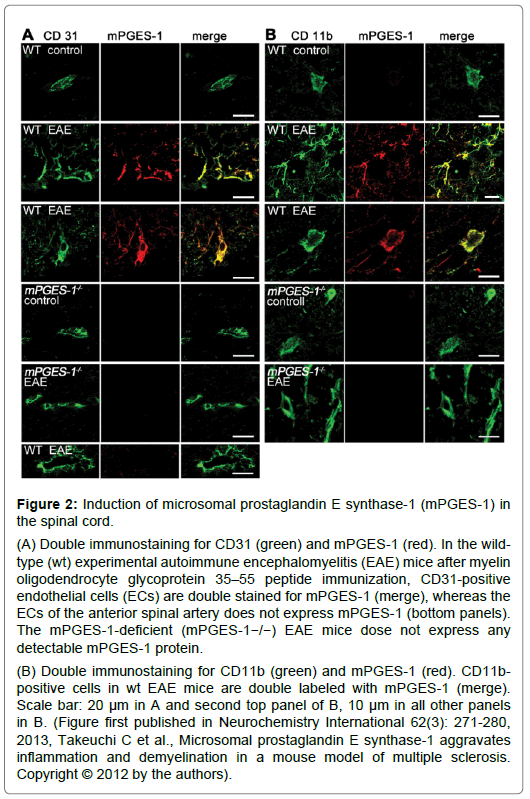
Figure 2: Induction of microsomal prostaglandin E synthase-1 (mPGES-1) in the spinal cord.
(A) Double immunostaining for CD31 (green) and mPGES-1 (red). In the wildtype (wt) experimental autoimmune encephalomyelitis (EAE) mice after myelin oligodendrocyte glycoprotein 35–55 peptide immunization, CD31-positive endothelial cells (ECs) are double stained for mPGES-1 (merge), whereas the ECs of the anterior spinal artery does not express mPGES-1 (bottom panels). The mPGES-1-deficient (mPGES-1−/−) EAE mice dose not express any detectable mPGES-1 protein.
(B) Double immunostaining for CD11b (green) and mPGES-1 (red). CD11bpositive cells in wt EAE mice are double labeled with mPGES-1 (merge). Scale bar: 20 μm in A and second top panel of B, 10 μm in all other panels in B. (Figure first published in Neurochemistry International 62(3): 271-280, 2013, Takeuchi C et al., Microsomal prostaglandin E synthase-1 aggravates inflammation and demyelination in a mouse model of multiple sclerosis. Copyright © 2012 by the authors).
In the brain and spinal cord, mPGES-1 activates several neuroinflammatory processes in neurodegenerative disease states and under physiological conditions. For example, mPGES-1 exacerbates hippocampal neuron injury induced by seizure and cerebral ischemia after transient occlusion of the hemilateral middle cerebral artery [4,26-29]. Moreover, mPGES-1 expression is associated with the β-amyloid plaque density, microglial accumulation, and learning impairment in subjects with Alzheimer’s disease [30]. The expression of mPGES-1 is also induced by lipopolysaccharide treatment and plays a crucial role in amyotrophic lateral sclerosis [3,31-34]. Finally, mPGES-1 stimulates macrophage activation in the spinal cord and is related with anoxia and the maintenance of wakefulness. According to these reports, PGE2 synthesized by mPGES-1 is a pathological mediator of and therapeutic target for neurodegenerative diseases and CNS injury [35-37].
Several studies have investigated the effects of PGE2 inhibition on EAE. COX-2 inhibitors limit the extent of EAE severity and T cell responses [13-15]. The administration of COX-2 inhibitors starting on the day of immunization are more effective at reducing EAE severity than administration starting 8 days or 14 days after immunization. Four subtypes of PGE2 receptors have been identified (E-prostanoid (EP) receptors EP1–4), and EP4-deficient mice show a significant attenuation of EAE symptoms. Moreover, the treatment of EP2-deficient mice with an EP4 antagonist from days 3 to 7 after immunization completely blocks the onset of EAE [13,15,38]. In addition, mPGES-1-deficient (mPGES-1-/-) mice show a delayed, brief period of symptomatic disease followed by recovery rather than progression, and exhibit the impairment for 26 days or 28 days after immunization (Figure 1B). Compared to wild-type (wt) mice, mPGES-1-/- mice exhibit a delayed EAE onset [11,12,18]. Moreover, mPGES-1 aggravates the inflammation and demyelination associated with EAE (Table 1 and Figure 3). Thus, PGE2 synthesized by COX-2 and mPGES-1 plays important roles in early disease onset and later during disease maintenance. Because PGE2 is a generalized production, the source of PGE2 is not limited in spinal cord and brain in acute inflammation of EAE.
| WT EAE (n=7) | mPGES-1-/- EAE (n=7) | p | |
|---|---|---|---|
| Inflammatory ratio* | 5.33 ± 4.86 | 0.24 ± 0.12 | <0.01 |
| Demyelinating score | 2.0 ± 0.81 | 0.1 ± 0.48 | <0.01 |
* Area of the inflammatory lesion / spinal cord area (%)
Table 1: The pathological analysis during the acute phase of EAE (19 days after immunization).
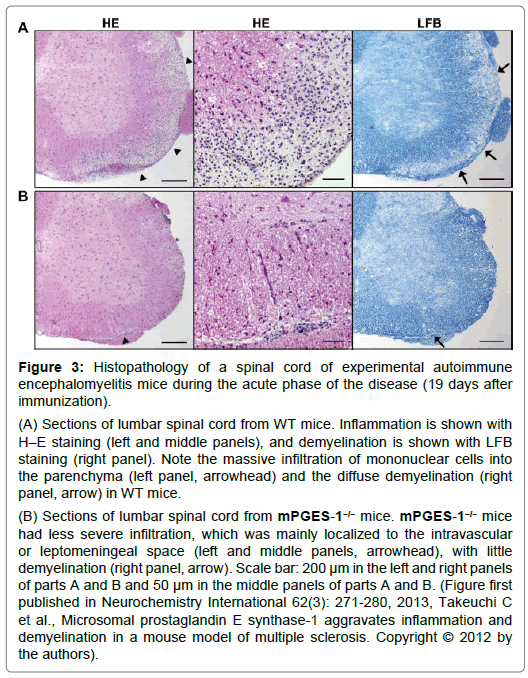
Figure 3: Histopathology of a spinal cord of experimental autoimmune encephalomyelitis mice during the acute phase of the disease (19 days after immunization).
(A) Sections of lumbar spinal cord from WT mice. Inflammation is shown with H–E staining (left and middle panels), and demyelination is shown with LFB staining (right panel). Note the massive infiltration of mononuclear cells into the parenchyma (left panel, arrowhead) and the diffuse demyelination (right panel, arrow) in WT mice.
(B) Sections of lumbar spinal cord from mPGES-1−/− mice. mPGES-1−/− mice had less severe infiltration, which was mainly localized to the intravascular or leptomeningeal space (left and middle panels, arrowhead), with little demyelination (right panel, arrow). Scale bar: 200 μm in the left and right panels of parts A and B and 50 μm in the middle panels of parts A and B. (Figure first published in Neurochemistry International 62(3): 271-280, 2013, Takeuchi C et al., Microsomal prostaglandin E synthase-1 aggravates inflammation and demyelination in a mouse model of multiple sclerosis. Copyright © 2012 by the authors).
However, researchers have not clearly determined the source of PGE2: VECs in CNS and mac/mic in the spinal cord or peripheral blood monocytes. Interestingly, based on the findings from bone marrow transplantation models, which enable EP expression or PGE2 synthesis to be blocked in peripheral immune cells, EP4 or COX-2 deletion in bone marrow-derived cells causes a significant delay in the EAE onset, but the animals ultimately experience serious paralysis, similar to the controls. Moreover, the deletion reduces the number of T cells and levels of IL-6 and interleukin-17 (IL-17) in the blood [39]. Therefore, the peripheral PGE2 /EP4 pathway is very important during EAE onset but does not control the severity of EAE. Researchers have hypothesized that inducible PGE2 synthesized by COX-2 and mPGES-1 in VECs in CNS and mac/mic facilitates pathological changes in the spinal cord of EAE models that lead to symptom manifestation.
In a clinical study, MS patients presented significantly higher levels of IL-1β in the serum and cerebrospinal fluid than healthy control subjects [40]. IL-1β has consistently been shown to play an important role in EAE. The administration of recombinant IL-1RN to EAE animals delays the onset of the disease and decreases its severity. Moreover, a mutation in the IL-1R1 gene in mice show complete resistance to EAE [16,17]. IL-1β also regulates blood-brain barrier (BBB) permeability through endothelial IL-1R1 and induces BBB disruption. IL-1β also exacerbates neuronal inflammation and is showed at high levels in infiltrating macrophages [41-48]. The expression of the IL-1β mRNA is induced in peritoneal leukocytes right after the initial induction of EAE [49]. In addition, macrophages have a minor contribution to the total IL-1β production during EAE pathogenesis and CD4-positive (CD4+) T cells are thought to be a major source of IL-1β in EAE pathogenesis [44,48-50]. IL-1β levels are increased in activated CD4+ T cells in inflammatory lesion in the spinal cord of EAE animals, and the increased IL-1β levels are mediated by mPGES-1 (Figure 4). Moreover, IL-1R1 expression in CD4+ T cells in EAE models is also controlled by mPGES-1. IL-1R1 is expressed on IL-17-producing helper T cells (Th17), but not type 1 helper T cells (Th1), in EAE mice and early Th17 differentiation is regulated by IL-1β signaling through IL-1R1 [18,50-52].
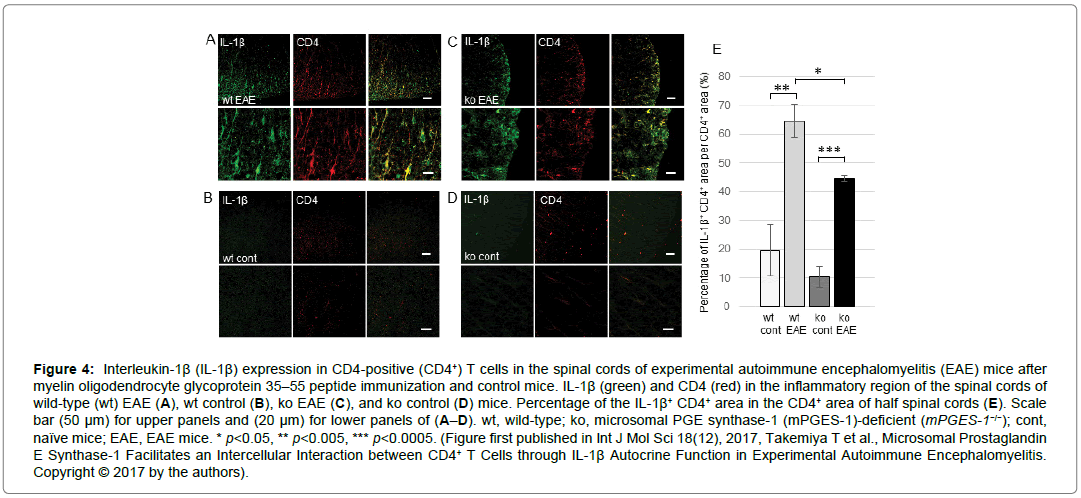
Figure 4: Interleukin-1β (IL-1β) expression in CD4-positive (CD4+) T cells in the spinal cords of experimental autoimmune encephalomyelitis (EAE) mice after myelin oligodendrocyte glycoprotein 35–55 peptide immunization and control mice. IL-1β (green) and CD4 (red) in the inflammatory region of the spinal cords of wild-type (wt) EAE (A), wt control (B), ko EAE (C), and ko control (D) mice. Percentage of the IL-1β+ CD4+ area in the CD4+ area of half spinal cords (E). Scale bar (50 μm) for upper panels and (20 μm) for lower panels of (A–D). wt, wild-type; ko, microsomal PGE synthase-1 (mPGES-1)-deficient (mPGES-1−/−); cont, naïve mice; EAE, EAE mice. * p<0.05, ** p<0.005, *** p<0.0005. (Figure first published in Int J Mol Sci 18(12), 2017, Takemiya T et al., Microsomal Prostaglandin E Synthase-1 Facilitates an Intercellular Interaction between CD4+ T Cells through IL-1β Autocrine Function in Experimental Autoimmune Encephalomyelitis. Copyright © 2017 by the authors).
IL-1β also stimulates IL-1R1 expressed on ECs and induces the production of IL-6 and chemokine ligand 2 in inflammatory lesions in EAE models whereas, no studies have evaluated the IL-1β production in ECs. Recently, we investigated whether IL-1β expression was increased in ECs by PGE2 synthesized from mPGES-1. When we compared IL-1β levels in ECs from wt and mPGES-1-/- mice, IL-1β levels were significantly elevated in wt EAE mice than in mPGES-1-/- EAE mice (Figure 5). Therefore, IL-1β also plays an important role in ECs, and IL-1β expression is regulated by PGE2 during EAE [53,44].
COX-2 expression is facilitated by IL-1β. IL-1β induces COX-2 expression in synovial fibroblasts and an intraperitoneal IL-1β injection induces the COX-2 mRNA expression in VECs in the brain [54-55]. The main protein that upregulates the COX-2 expression in the spinal cord is IL-1β, which promotes PGE2 production and contributes to pain hypersensitivity. IL-1β also plays a role on mediating the activity of nuclear factor kappa B and the COX-2 transcription in cells of the BBB in response to inflammation. COX-2 inhibitors prevent IL-1β- induced increases in PGE2 production in the brain. It suggests that IL-1β controls PGE2 production by modulating the synthesis of COX- 2. Moreover, crosstalk between microvascular ECs and tumor cells increases COX-2 and mPGES-1, which are strongly inhibited by an IL-1R antagonist [54-59]. Thus, IL-1β plays an important role in the induction of COX-2 and mPGES-1 to produce the pathophysiological protein PGE2. In contrast, IL-1β levels in CD4+ T cells were elevated by PGE2 derived from mPGES-1 in EAE spinal cords (Figure 4) [18]. Moreover, IL-1β was also increased in ECs present in inflammatory lesions in EAE models therefore, IL-1β expression in CD4+ T cells and ECs is stimulated by PGE2 in the spinal cord of EAE animals (Figure 5) [53].
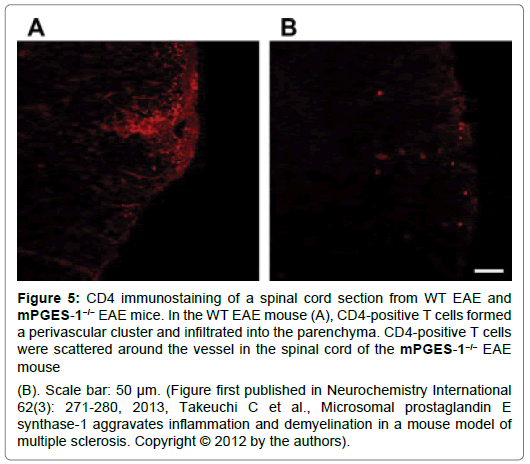
Figure 5: CD4 immunostaining of a spinal cord section from WT EAE and mPGES-1−/− EAE mice. In the WT EAE mouse
(A), CD4-positive T cells formed a perivascular cluster and infiltrated into the parenchyma. CD4-positive T cells were scattered around the vessel in the spinal cord of the mPGES-1−/− EAE mouse
(B). Scale bar: 50 μm. (Figure first published in Neurochemistry International 62(3): 271-280, 2013, Takeuchi C et al., Microsomal prostaglandin E synthase-1 aggravates inflammation and demyelination in a mouse model of multiple sclerosis. Copyright © 2012 by the authors).
PGE2 also regulates T cell activation and differentiation, depending on the cellular environment. PGE2 enhances the Th17 responses of CD4+ T cells via EP4 and EP2 promotes Th17 differentiation and function through EP4 and EP2 and works with IL-23 and IL-1β to enhance IL-17A expression through EP4 [60-63].
In wt EAE mice, CD4+ T cells form perivascular clusters and infiltrate the spinal cord parenchyma, whereas they are scattered around vessels in the spinal cord of mPGES-1-/- EAE mice and no T cells are detected in the spinal cords of both control mice Researchers have postulated that CD4+ T cells infiltrate the spinal cord of animals with EAE through the disrupted blood-spinal cord barrier, and the infiltration and/or the activation of CD4+ T cells is regulated by PGE2. Because CD4+ T cells almost completely colocalize with IL-1β in wt EAE mice, whereas IL-1β is expressed at very low levels in the spinal cord of mPGES-1-/- EAE mice, this pathway may represent the mechanism by which PGE2 directly activates CD4+ T cells to increase IL-1β production in EAE models [12,18] (Figure 6).
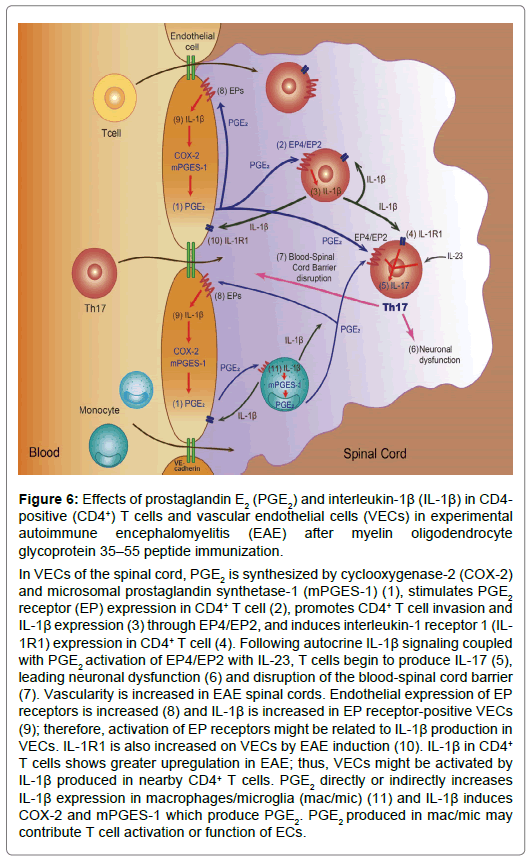
Figure 6: Effects of prostaglandin E2 (PGE2) and interleukin-1β (IL-1β) in CD4- positive (CD4+) T cells and vascular endothelial cells (VECs) in experimental autoimmune encephalomyelitis (EAE) after myelin oligodendrocyte glycoprotein 35–55 peptide immunization.
In VECs of the spinal cord, PGE2 is synthesized by cyclooxygenase-2 (COX-2) and microsomal prostaglandin synthetase-1 (mPGES-1) (1), stimulates PGE2 receptor (EP) expression in CD4+ T cell (2), promotes CD4+ T cell invasion and IL-1β expression (3) through EP4/EP2, and induces interleukin-1 receptor 1 (IL- 1R1) expression in CD4+ T cell (4). Following autocrine IL-1β signaling coupled with PGE2 activation of EP4/EP2 with IL-23, T cells begin to produce IL-17 (5), leading neuronal dysfunction (6) and disruption of the blood-spinal cord barrier (7). Vascularity is increased in EAE spinal cords. Endothelial expression of EP receptors is increased (8) and IL-1β is increased in EP receptor-positive VECs (9); therefore, activation of EP receptors might be related to IL-1β production in VECs. IL-1R1 is also increased on VECs by EAE induction (10). IL-1β in CD4+ T cells shows greater upregulation in EAE; thus, VECs might be activated by IL-1β produced in nearby CD4+ T cells. PGE2 directly or indirectly increases IL-1β expression in macrophages/microglia (mac/mic) (11) and IL-1β induces COX-2 and mPGES-1 which produce PGE2. PGE2 produced in mac/mic may contribute T cell activation or function of ECs.
An analysis of Th17 and Th1 cells in culture supernatants revealed high IL-17 expression in Th17 cells and high IFN-γ expression in Th1 cells in EAE models [64]. IL-17 staining is colocalized with CD4+ T cells in wt EAE mice, whereas morphological changes are restricted and IL-17 staining is weak in mPGES-1-/- EAE mice. In contrast, IFN-γ is co-expressed in few CD4+ T cells in either wt or mPGES-1-/- EAE mice [18]. Thus, mPGES-1 stimulates the production of IL-17 in CD4+ T cells. In contrast, interferon-γ (IFN-γ) staining is partially colocalized with CD4+ T cells in both wt and mPGES-1−/− EAE mice, suggesting that PGE2 does not regulate the production of IFN-γ, which means an activity of Th1. The EAE incidence and scores are reduced when EAE mice are mediated with anti-IL-17 antibodies prior to the observed increase in CD4+ IL-17+ T cells, suggesting that CD4+ T cells control tissue inflammation by inducing IL-17 production [65]. Because PGE2 regulates IL-17 expression in EAE models, we postulate that this mechanism is a potential explanation for the participation of PGE2 in EAE pathology.
Inflammation induces the vasodilation of small blood vessels, subsequent more perfusion and resulting in an obvious increase in vessel density, which is known as vascularity. After EAE induction, vascularity is caused by vasodilation and angiogenesis. Angiogenesis is facilitated by vascular endothelial growth factor (VEGF), which leads to the degeneration of the vascular basement membrane and BBB breakdown [66,67]. In MS patients, BBB dysregulation and the transendothelial migration of activated leukocytes are the earliest signs of cerebrovascular dysfunction. VEGF is involved in EAE process during the acute phase [66,68,69]. In addition, inhibition of VEGF receptor 2 reduces clinical signs of EAE in the acute phase of the EAE progression. Furthermore, the number of blood vessels increases during the relapse phase of EAE animals [66,69]. Thus, angiogenesis aggravates inflammation in the spinal cord of EAE animal [70,71]. Vascularity is triggered upon EAE induction, regardless of the presence of mPGES-1. In addition, the PGE2 produced by mPGES-1 stimulates vascularity in the spinal cord of animal models of EAE, and aggravates inflammation, demyelination, and paralysis. Therefore, vascularity is one mechanism by which PGE2 aggravates EAE. Furthermore, IL-1β expression is significantly increased in ECs from EAE mice, and the increase in IL- 1β levels is mediated by mPGES-1 (Figure 5) [12,18,53]. Furthermore, endothelial IL-1β stimulates COX-2 and mPGES-1 expression to produce PGE2; therefore, the IL-1β- PGE2 autocrine signaling pathway in ECs stimulates vascularity. In contrast, the induction of angiogenesis after inflammation promotes neuronal remodeling through the production of prostaglandin I2 during the EAE chronic phase [70,71].
IL-17 is a proinflammatory cytokine produced by activated memory T cells, and the IL-17s are secreted proteins of 150–180 amino acids. There are at least six members of the IL-17 family (IL-17 (IL-17A), IL- 17B, IL-17C, IL-17D, IL25(IL-17E and IL-17F) in mice and humans [72,73]. The receptor for IL-17 (IL-17R) is a transmembrane protein of approximately 130 kDa. The IL-17 is expressed only by T-cells, whereas its receptor is expressed in all tissues. Moreover, four receptors are identified which share partial sequence homology to IL-17R. EAE development is suppressed in IL-17 knockout mice. The severity of EAE in mice immunized by PLP is reduced when IL-17 is neutralized in vivo, demonstrating the crucial role of IL-17 in EAE induction [74-76]. In addition, T cell-intrinsic apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) is required for the effector stage of EAE, and ASC deficiency in T cells impaired TH17- but not TH1-mediated EAE. Moreover, IL-1β is produced by a TH17 cellintrinsic ASC-NOD-like receptor 3-Caspase-8 inflammasome during CNS inflammation. There is an autocrine action of TH17-derived IL- 1β through IL-1R [50]. We found that PGE2 facilitates this autocrine function of IL-1β [18]. It is clear that IL-17 produced in Th17 cells has an important role on EAE. In contrast, IL-17 is also produced in CD8+ T cell, γδT cells, neutrophil or monocyte, therefore in the future we need to investigate the role and the regulatory mechanisms of IL-17 produced by other cells [77].
Next, I discuss other cytokines concerning with EAE and Th17. IL- 12p40 knockout mice show no clinical symptoms with no inflammation, whereas IL-12p35 knockout mice show more severe symptoms with severe inflammation. It suggests that p40-containing cytokine distinct from IL-12 is essential for the development of EAE. Moreover, IL-23 is a heterodimeric cytokine composed of a p19 subunit and the p40 subunit of IL-12, and IL-23p19 knockout mice are resistant to EAE. IL-23 also promotes an activation of distinct CD4 T cell producing IL-17. In addition, mice whose T cells cannot respond to signaling of TGF-β lack Th17 cells and do not develop EAE. Local administration of antibody to block TGF-β prevents the differentiation of Th17 cell and the onset of EAE [78-82]. On the contrary, mice whose T cells overexpress TGF-β develop more severe EAE and the T cells produce massive amounts of IL-17. These reports suggest that TGF-β regulates the differentiation of Th17 cell. Furthermore, IL-6 knockout mice do not develop a TH17 response. The orphan nuclear receptor RORγt expression is induced by IL-6 and TGF-b and RORγt induces transcription of the genes encoding IL-17 [83-85]. Moreover, IL-21 potently induces TH17 differentiation. IL-21 knockout mice impair the generation of TH17 cells and protect against EAE. IL-9 knockout mice are resistant to the induction of EAE and exhibit fewer inflammatory infiltrates in the CNS, with lower levels of IL-17 and IFN-λ expression. Th17 cells express IL-22, an IL-10 family member, at substantially higher amounts than Th1 or Th2 cells. Similar to IL-17, IL-22 expression is regulated by TGF in the context of IL-6 and other proinflammatory cytokines [86-88]. In addition, IL-27 suppresses the development of Th17 cells mediated by IL-6 and TGF-b, and IL-27 suppresses IL-6-mediated T cell proliferation. IFN-β treated animals show a decrease of IL-17 expression and IFN-β knockout mice exhibit earlier onset and more rapid progression in EAE. Furthermore, CD4 T cells in Tyk2 knockout mice reduce the IL-17A level in response to MOG35–55 [89-93]. Many cytokines including IL-17 are related with Th17 differentiation; therefore Th17 is potent T cell to effect on inflammation and CNS injury in EAE and MS (Table 2).
In the EAE spinal cord, PGE2 derived from mPGES-1 promotes CD4+ T cell invasion and facilitates IL-1β production by CD4+ T cells via EP receptors; IL-1β in turn participates in autocrine signaling through IL-1R1 expressed on CD4+ T cells. PGE2 also increases vascularity and endothelial IL-1β expression [18,53]. Th17 cells are pathogenic in many autoimmune diseases, and the induction and expansion of Th17 cells is regulated by IL-23 and IL-1β [51] through IL-1R1. Furthermore, PGE2, IL-23, and IL-1β differentially participate in the regulation of CD4+ T cell Th1/Th17 immune responses. Th17 cells induce severe but localized and partial changes in neuronal intercellular calcium concentrations as an early sign of neuronal damage. IL-17 also affects inflammatory mediator production in a wide range of cells, including myeloid cells, ECs, and epithelial cells, to increase the levels of IL-6, IL-1β, MMP-3, and macrophage inflammatory protein-2; importantly, these factors induce neuronal dysfunction and BBB disruption in EAE models. Recently, a repulsive guidance molecule was identified in Th17 cells and shown to dephosphorylate Akt in a mechanism leading to neuronal death. These findings support the hypothesis that endothelial mPGES-1 derived PGE2 may be a key factor leading to IL- 1β production in activated CD4+ T cells and ECs and to subsequent IL-17 production in EAE models [94-98].
Finally, we must consider whether peripheral monocytes are a source of PGE2. Peripheral PGE2 produced by monocytes induces IL-6 release through EP4 expressed on monocytes, which in turn induces IL- 17 secretion from T cells. Moreover, PGE2 induces MMP-9 expression in T cells through EP4, leading to a disruption of the blood-spinal cord barrier. Furthermore, COX-2/PGE2/EP4 signaling in peripheral immune cells facilitates the development of EAE, particularly during disease onset; however, EAE severity and maintenance/progression are not affected by peripheral immune cells but instead appear to be regulated by mPGES-1/PGE2/IL-1β signalling between ECs and CD4+ T cells in the spinal cord. Accordingly, CNS and peripheral PGE2 participate in different aspects of EAE pathology [39].
PGE2 induces the EAE progression by regulating IL-1β production and subsequently promoting neuronal dysfunction and disruption of the BBB in the spinal cord of subjects with EAE, leading to serious EAE symptoms. Furthermore, intercellular signalling pathways including PGE2 and IL-1β in the spinal cord may be an important therapeutic target in MS.
This work was supported by a grant from KAKENHI (17K10064). The author would like to thank the staff of the Medical Research Institute at Tokyo Women’s Medical University for their assistance with my research.
